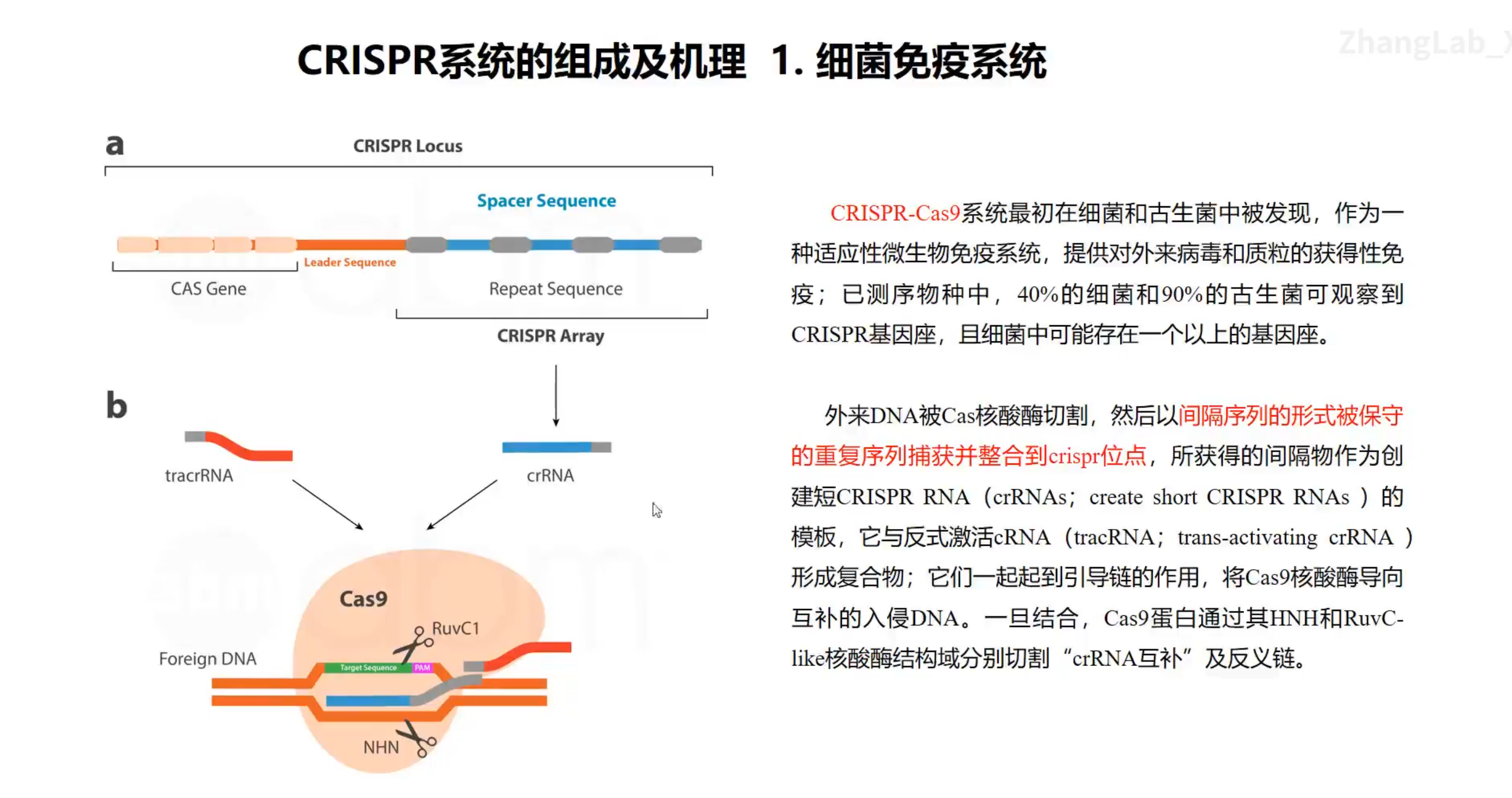
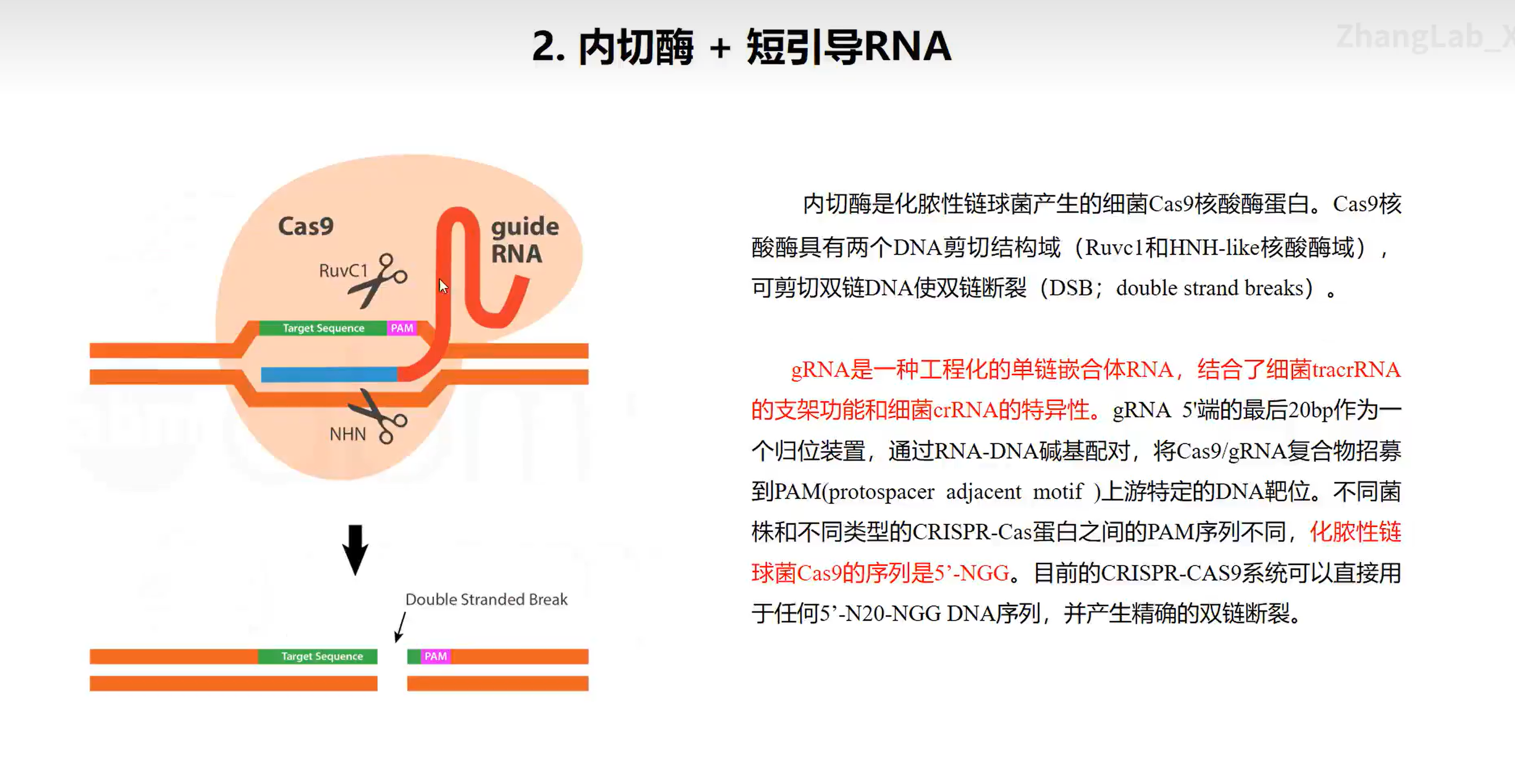


原理示意图

BsmBI识别位点



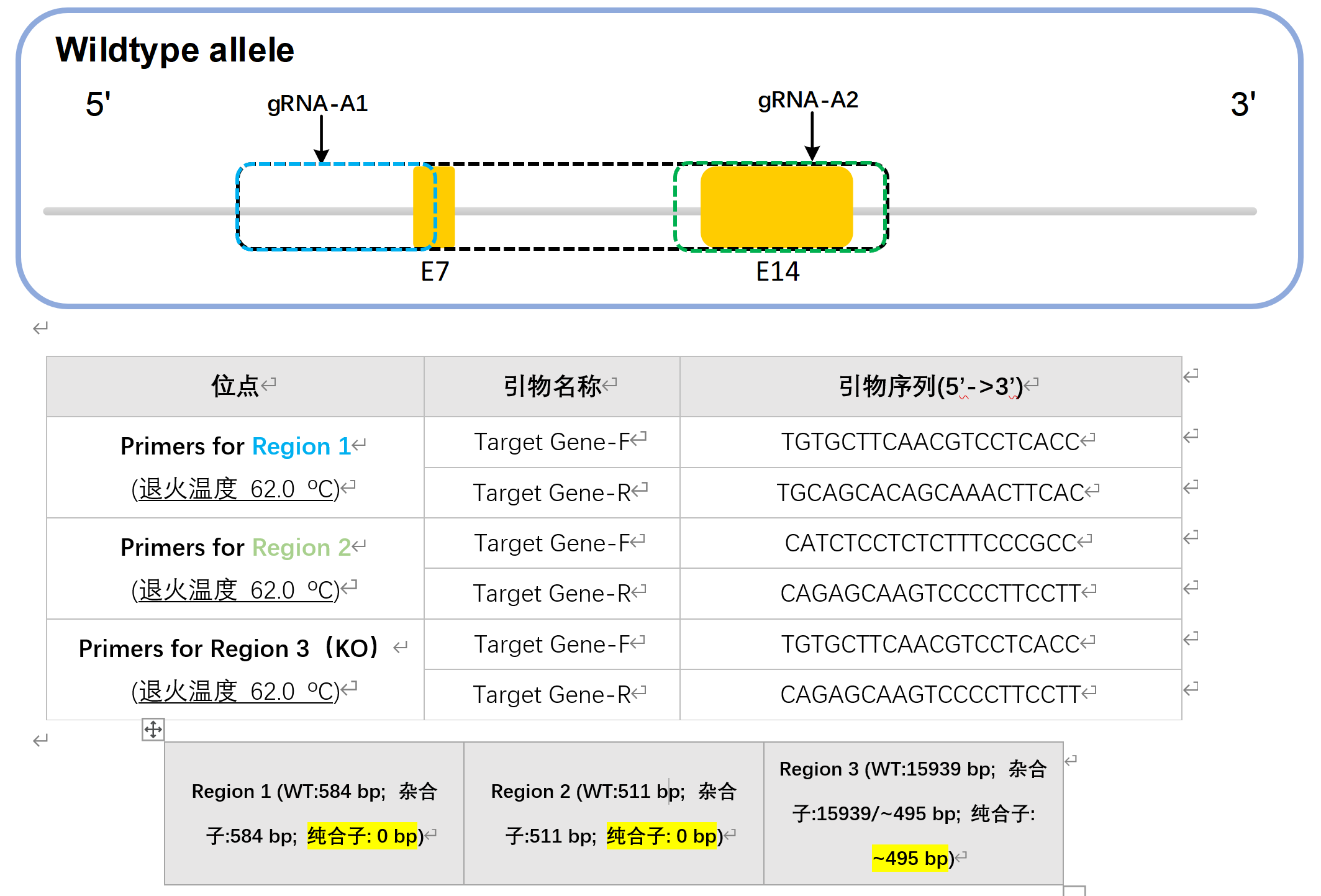
引物设计

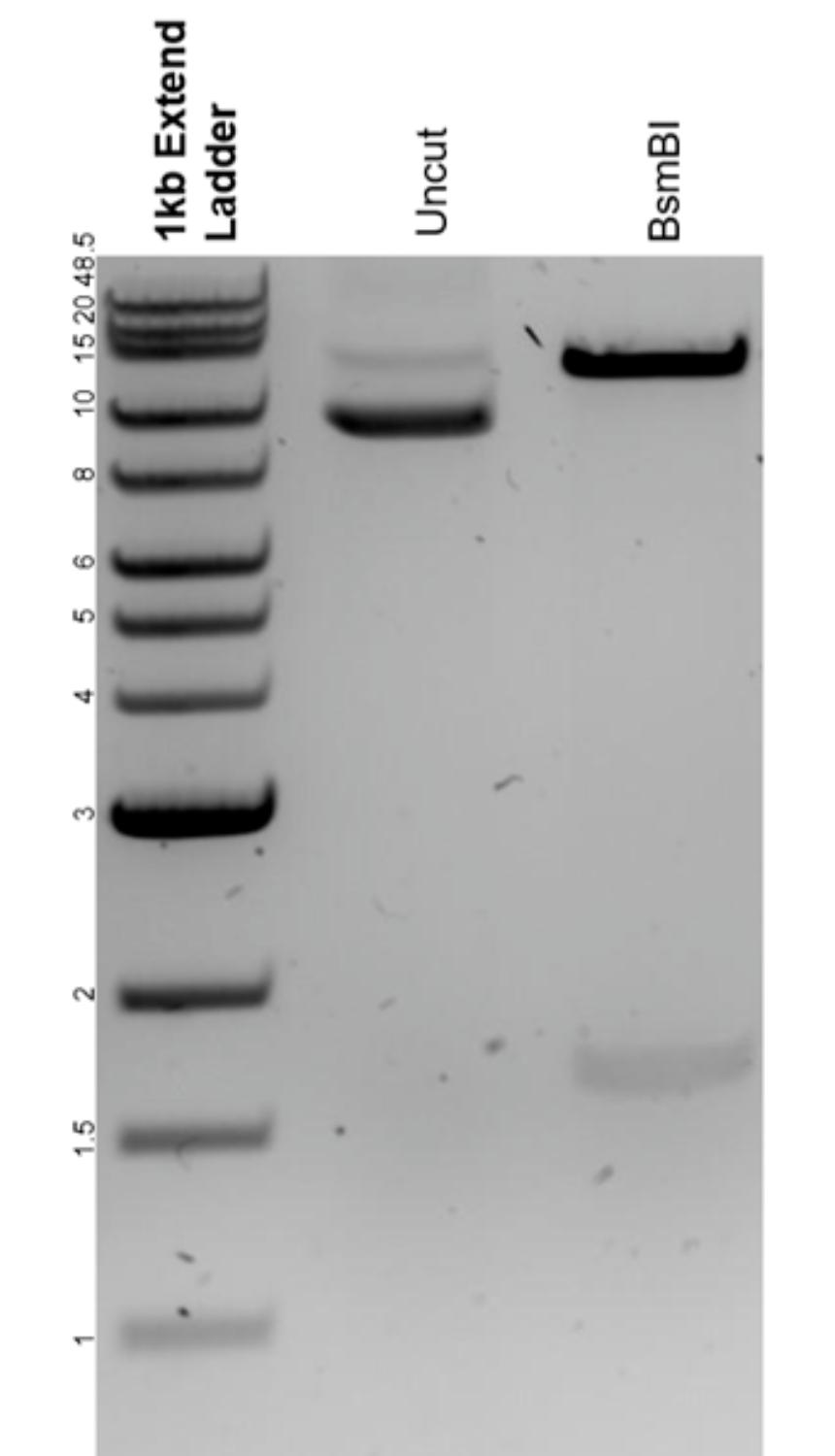
酶切后的线性化载体应比环状载体跑得慢
| Component | 5微升体系用量 | 20微升体系用量 |
|---|---|---|
| T4 DNA ligase (BioLabs, M0202L) | 0.25 μL | 1 μL |
| T4 DNA ligase reaction buffer (10X) | 0.5 μL | 2 μL |
| digested LentiCRISPRv2 plasmid | 4 μL(32ng) | 50ng(0.02pmol) |
| 稀释后的Duplex | 1 μL (0.015pmol) | 37.5ng (0.06pmol) |
| Nuclease-free water | to 5 μL | to 20 μL |
| total | 5 μL | 20 μL |