

Fig4F. Multi-omic profiling reveals age-related immune dynamics in healthy adults

De novo gene synthesis by an antiviral reverse transcriptase. Fig1B

METTL17 is an Fe-S cluster checkpoint for mitochondrial translation. Fig1B/D

The local microenvironment drives activation of neutrophils in human brain tumors. Fig3G

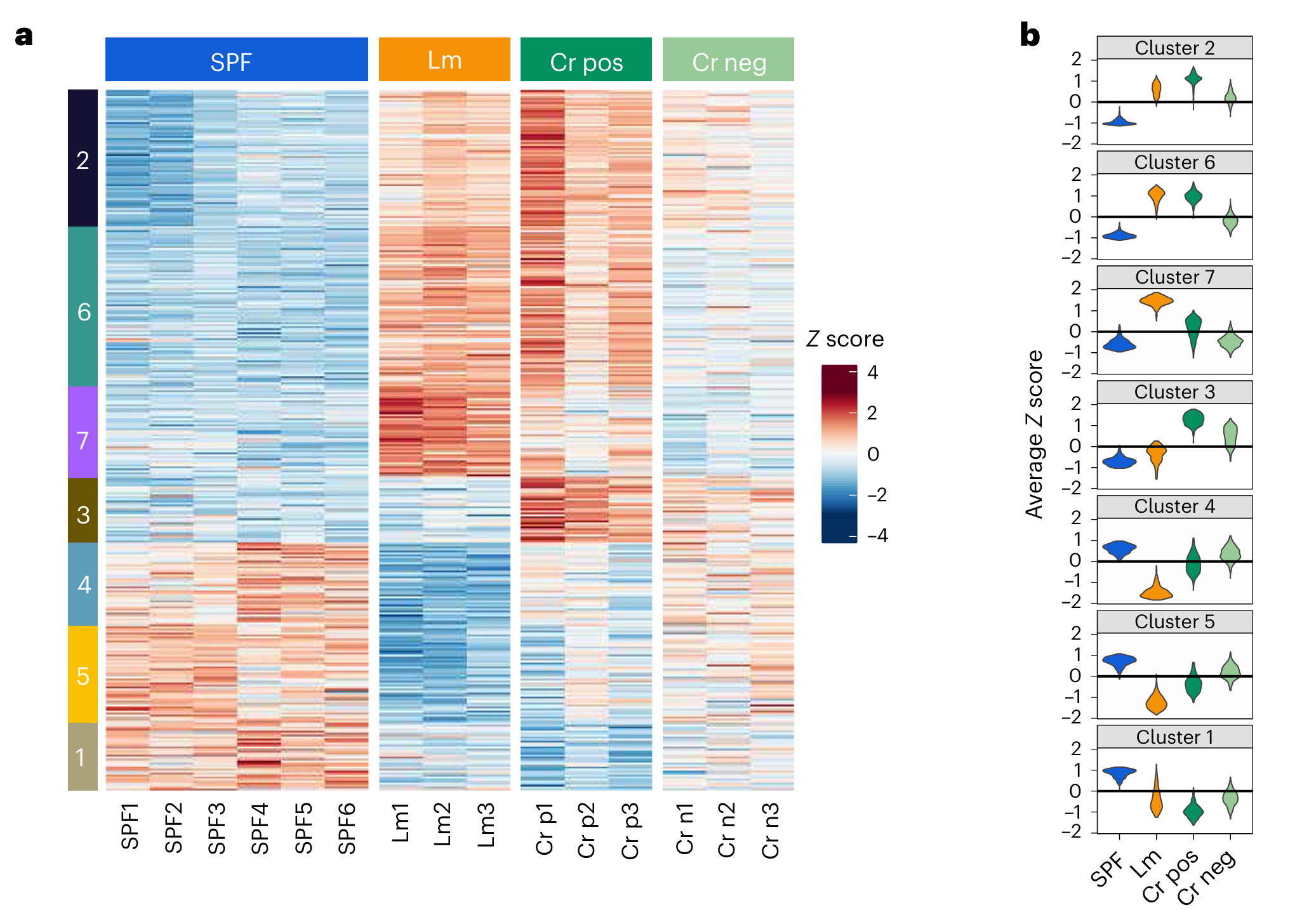
Fig3. Enteric bacterial infection stimulates remodelling of bile metabolites to promote intestinal homeostasis

Identification of markers correlating with mitochondrial function in myocardial infarction by bioinformatics

The local microenvironment drives activation of neutrophils in human brain tumors. Fig2B

The local microenvironment drives activation of neutrophils in human brain tumors. Fig2H


Small extracellular vesicles from young plasma reverse age-related functional declines by improving mitochondrial energy metabolism. Fig3D



Obesity reshapes regulatory T cells in the visceral adipose tissue by disrupting cellular cholesterol homeostasis. Fig1A

Y chromosome loss in cancer drives growth by evasion of adaptive immunity. Fig4A