# 免疫共沉淀 (Co-IP)
## 实验原理
免疫沉淀 (Immunoprecipitation, IP ) 是指利用抗原抗体特异性反应从细胞/组织/血液混合物中富集纯化其特异性的靶标或抗原。
免疫共沉淀( Co-IP ) 指用抗体用于从混合样品中富集纯化其靶抗原及其结合蛋白。在这种情况下,抗原是诱饵蛋白,而其结合蛋白是通过抗体--抗原相互作用共同纯化的相互作用的蛋白。
通常利用Co-IP实验可以:
(1)测定两种甚至更多种蛋白质是否在体内结合;
(2)鉴定一种特定蛋白质的作用搭档;
(3)分离得到天然状态的相互作用蛋白复合物。
Co-IP大致步骤:首先抗体对抗原(目的蛋白)进行特异性结合, 接着通过偶联在微珠上的亲和蛋白(如Protein A sepharose beads)对抗体Fc 端的结合形成“beads- 抗体- 目的蛋白”三联体,经洗涤去除未 结合的杂蛋白,然后SDS sample buffer 或酸性洗脱的方式使抗体、目的蛋白一起脱落下来,最后经 Western Blot 检测,胶片显影或仪器成像,观 察是否有目的蛋白带,来判断抗体是否成功捕获了目的蛋白。
 ## 名词解释
* **IB:**即immunoblotting(免疫印迹)也就是常规的Western Blotting,用于检测目的蛋白。
* **IP:**即immunoprecipitation(免疫沉淀)这一步主要是为了纯化富集目的蛋白。
* **Input:**全细胞裂解液,可认为是阳性组。也就是处理完样本得到的细胞样品,在做IP实验之前,需要预留Input,在检测IP的结果同时确认样本中含有蛋白A和蛋白B。
* **Anti-A:**A蛋白的免疫共沉淀抗体。
* **IgG:**Anti-A的同型对照抗体,即跟Anti-A同种属来源,如Anti-A是鼠源IgG型,则同型对照抗体需要选择Mouse IgG。
## 对照设置
* Co-IP(免疫共沉淀)一般分为内源性和外源性,我们可以简单理解为内源性CO-IP是检测细胞本身含有的蛋白A和蛋白B互作有无;外源性CO-IP是将蛋白A和蛋白B在细胞内过表达后,再检测二者互作有无。
对于内源性Co-IP实验,如果出现阴性结果,也有可能是蛋白在细胞内表达过低导致,这种情况也可以先做过表达Co-IP作为对照。只有设置了正确的对照,出来的结果才是可以被合理解释的,也是被认可的。
### 1、内源性Co-IP检测
对于Protein A/G微珠,一般会设置Input和同型对照:Input作为**阳性对照**,无需进行免疫沉淀步骤,直接使用制备好的样本进行检测,以证明靶蛋白存在于样本中;同型抗体作为**阴性对照**,使用同种属/类型的无关IgG进行IP,体现蛋白与IgG结合的背景信号,以排除假阳性结果。
## 名词解释
* **IB:**即immunoblotting(免疫印迹)也就是常规的Western Blotting,用于检测目的蛋白。
* **IP:**即immunoprecipitation(免疫沉淀)这一步主要是为了纯化富集目的蛋白。
* **Input:**全细胞裂解液,可认为是阳性组。也就是处理完样本得到的细胞样品,在做IP实验之前,需要预留Input,在检测IP的结果同时确认样本中含有蛋白A和蛋白B。
* **Anti-A:**A蛋白的免疫共沉淀抗体。
* **IgG:**Anti-A的同型对照抗体,即跟Anti-A同种属来源,如Anti-A是鼠源IgG型,则同型对照抗体需要选择Mouse IgG。
## 对照设置
* Co-IP(免疫共沉淀)一般分为内源性和外源性,我们可以简单理解为内源性CO-IP是检测细胞本身含有的蛋白A和蛋白B互作有无;外源性CO-IP是将蛋白A和蛋白B在细胞内过表达后,再检测二者互作有无。
对于内源性Co-IP实验,如果出现阴性结果,也有可能是蛋白在细胞内表达过低导致,这种情况也可以先做过表达Co-IP作为对照。只有设置了正确的对照,出来的结果才是可以被合理解释的,也是被认可的。
### 1、内源性Co-IP检测
对于Protein A/G微珠,一般会设置Input和同型对照:Input作为**阳性对照**,无需进行免疫沉淀步骤,直接使用制备好的样本进行检测,以证明靶蛋白存在于样本中;同型抗体作为**阴性对照**,使用同种属/类型的无关IgG进行IP,体现蛋白与IgG结合的背景信号,以排除假阳性结果。
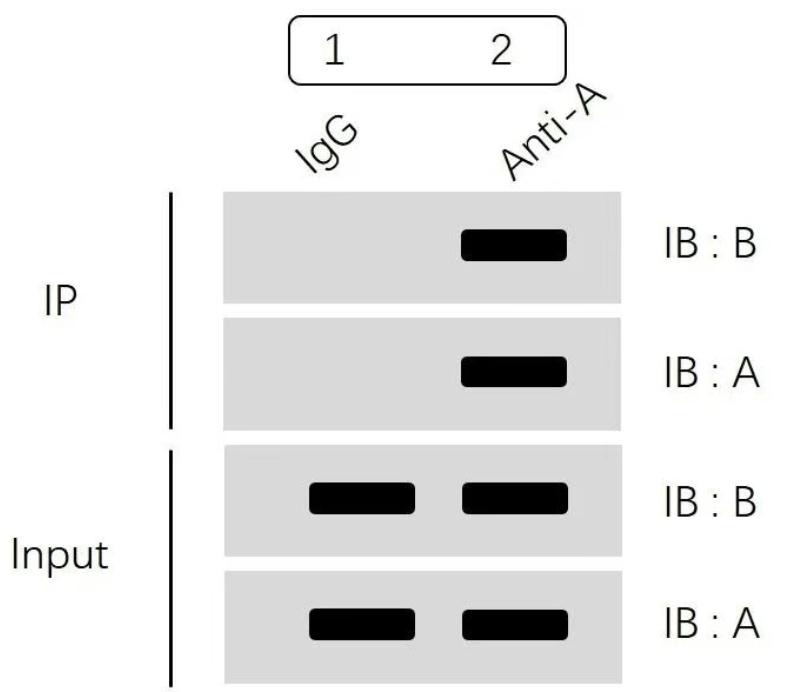
内源性CO-IP检测
**上面示意图分为两大部分**:IP 部分和 Input 部分。
* 首先看Input 部分,通过分别检测蛋白A和蛋白B表达水平可以确认:1组(IgG组)和2组(Anti-A组)中蛋白A、蛋白B均有表达,并且表达量一致,排除了因部分组别部分蛋白表达缺失、不同组蛋白表达量差异化大等原因造成的假阴性或者假阳性。
* 然后再看IP部分:“IB:A”结果显示1组(IgG组)不结合蛋白A ,2组(Anti-A组)可以特异性富集蛋白A。“IB:B”结果显示我们IgG组没有非特异性结合,Anti-A组通过特异性富集蛋白A进而拉下蛋白B,即提示我们蛋白A和蛋白B可形成复合物,存在互作。

另一种表示方式
也可以用另一种分组方式。其中IP组又分为IgG组(阴性对照组)和实验组,并通过WB去验证蛋白A和B在每一个组里面是否存在。
* 由Input组得到结论:蛋白A与蛋白B都是存在的,证明样本处理提取蛋白的步骤没有任何问题。这也提示:本实验一般是选取同一份细胞样品,提前取出部分做Input,其余均分为两份即IgG组(阴性对照组)和实验组。
* IP部分:2组(IgG组)结果显示,蛋白A与蛋白B均没有条带,说明利用IgG抗体没有把蛋白A和B沉淀下来,证明蛋白A和B与IgG没有结合,同时排除了蛋白与抗体非特异性结合的可能性;而实验组蛋白A和B均有条带,表示利用蛋白A的抗体进行免疫沉淀,富集了蛋白A,同时蛋白B也被拉下来,即提示我们蛋白A和蛋白B可形成复合物,存在互作。
### 2、外源性Co-IP检测
针对标签蛋白的转染过表达的IP/Co-IP,其阴性对照一般选择未转染该标签细胞制备的lysate,以体现不含标签的蛋白与IP捕获抗体结合的背景信号,排除IP假阳性结果。

过表达一种蛋白

过表达2种蛋白
**结果同样分为两大部分**:IP 部分和 Input 部分。
* 首先看Input 部分,通过分别检测HA和Flag表达水平,我们可以确认:A-Flag和B-HA过表达后都能被检测到,并且表达量一致,排除了因样品量不均匀、样品组分缺失等原因造成的假阴性或者假阳性。
* 然后我们再看IP部分,“IB:Flag ”结果显示1、3组通过IP:Flag可以特异性识别Flag标签并富集蛋白A。“IB:HA”结果显示1、2对照两组没有捕获蛋白B,第3组通过特异性富集蛋白A进而捕获蛋白B,即提示我们蛋白A和蛋白B可形成复合物,存在互作。
## WB检测抗体及抗体的选择
 如捕获标签-融合蛋白,可以选用纳米抗体,即羊驼重链抗体可变区(VHH),使用普通二抗也可以避免轻重链干扰。
## 实验步骤
### 前期准备
* 在2个T75或者2个10cm皿内铺板293T,使用PEI转染质粒,等待2天;
### Day1:收集、裂解细胞
* 预冷PBS、EP管,取冰若干;
* 提前配置蛋白裂解液:每组样本准备1000μL的RIPA(中)+10μL的PMSF(100×)/100μL的PPI(10×),置于冰上。
* 吸出两个瓶/皿中的培养液,加1mL预冷的1×PBS,再用细胞刮刀将细胞刮下,将细胞转入预冷的EP管中,4℃,500G离心5min,弃去上清,用预冷的1X PBS重悬细胞,离心,弃上清,重复两次。
* 使用1000μL裂解液重悬细胞。
* 将EP管放于冰上30min,每隔10min重悬细胞一次。
* 摩天轮 4°C 转动 40-60min 以彻底裂解细胞(摩天轮上ep管可以用封口膜封口);
* 将蛋白冻存于-80℃,或直接进行下一步;
### Day2:平衡磁珠、结合蛋白
### 【康体FLAG磁珠】
* 取出蛋白,在冰上融化,12000G离心15min;
* 吸出50μL作为input,向其中加入12μL 5×SDS,混匀,置于-80℃;
* 配置1×PBST:1×PBS中加入0.05%的Tween20(50mL:25μL),配置完后置于冰上;
* 涡旋Anti-Flag Magarose Beads磁珠,吸取25μL置于1.5mL 无酶EP管中;
* 加入500μL 1XPBST(1X PBS+0.05%Tween-20),手动摇晃60s;
* 放入磁力架静置60s,吸去上清,再加入500μL 1XPBST,随后在4℃摩天轮转动5min;再次放入磁力架吸附,重复3次;
* 在平衡好的磁珠中加入细胞蛋白提取液,用封口膜封口后4℃摇床翻转过夜;
{% file src="" %}
### 【Thermo FLAG磁珠】
* 准备工作:
* 打开95℃金属浴;
* 准备RIPA(中)、 5×SDS、1×PBS以及10×PPI,以及若干EP管,置于冰上;
* 取出蛋白样品,在冰上融化,12000G离心15min;
* 取出磁珠,使用前将磁珠平衡至室温;
* 离心期间平衡洗涤磁珠、标记无酶离心管:
* 配置含PPI的裂解液(Binding Buffer):RIPA(中)加入十分之一体积的10× PPI(10mL:1mL),配置完后置于冰上;
* 涡旋或吹匀Anti-Flag Magnetic Agarose磁性琼脂糖珠,为每个样品吸取25μL(说明书推荐50μL)置于1.5mL 无酶EP管中;
* 每管加入500μL 上述配置的裂解液,手动摇晃60s混匀;
* 放入磁力架静置60s,吸去上清,加入500μL上述裂解液重悬,随后在4℃摩天轮转动5min;
* 再次放入磁力架吸附,弃上清,加入500μL裂解液,重悬洗涤5min,再磁力架吸附,弃去上清;
* 从离心完的蛋白样品上清中吸出50μL作为input,向其中加入12μL 5×SDS,混匀,95℃加热10min,置于-80℃;
* 其余样品加入到平衡好的磁珠管中,再加入10× PPI,用封口膜封口后4℃摇床翻转过夜;
{% file src="" %}
### 【Thermo Protein A/G 磁珠】
* 准备工作:
* 提前打开95℃金属浴;
* 准备5× SDS以及若干EP管(蛋白上清若干管,input若干管,磁珠若干管);
* 取出蛋白样品,在冰上融化,12000G离心15min;
* 从离心完的蛋白样品上清中吸出50μL作为input,向其中加入12μL **5×SDS**,混匀,95℃加热10min,置于-80℃;
* 将其余**蛋白上清**分为2份,分别与约10μL的IgG同型对照抗体/目的蛋白**抗体**进行混合(每个T75的293T约1.2mg蛋白,抗体浓度一般1-5 µL/mg;2个T75的蛋白量即对应2-10μL抗体),**4℃翻转过夜**(若阴性对照有阳性结果,可以减少孵育时间至室温孵育1-2h);
{% file src="" %}
### Day3:洗涤、洗脱
### 【康体FLAG磁珠】
* 用磁力架静置分离磁珠1min,吸去上清液;
* 加入500μL PBST悬浮磁珠,4℃冰箱旋转摇晃5min;再静置,如此重复洗涤4次;
* 配置30μL 1× SDS loading buffer,加入PMSF/PPI;
* 吸去上清液,用30μL **1×SDS** loading重悬磁珠;
* 95℃加热磁珠10min,使免疫沉淀复合物与磁珠分离;
* 用磁力架静置分离磁珠,取上清,冻于-80℃,跑SDS-PAGE。
### 【Thermo FLAG磁珠】
* 准备工作:
* 提前打开95℃金属浴;
* 配置含PPI的1×PBS(Wash Buffer):1×PBS中加入十分之一体积的10× PPI(10mL:1mL),配置完后置于冰上;
* 配置含PPI的ddH2O:ddH2O中加入十分之一体积的10× PPI(2mL:200μL),配置完后置于冰上;
* 配置1× SDS PAGE sample buffer:5×SDS+10×PPI用RIPA(中)稀释;
* 用磁力架分离磁珠,吸弃上清,加入500μL 配置好的Wash Buffer,混匀,磁力架分离。重复洗一遍;
* 加500μL 配置好的ddH2O,混匀洗涤1遍;
* 用磁力架分离磁珠,吸弃上清,加入50μL(说明书推荐100μL)**1× SDS PAGE** sample buffer,混匀;
* 95-100℃加热10分钟(期间标记EP管);
* 用磁力架分离磁珠,吸取上清至新的EP管,-80℃保存。
### 【Thermo Protein A/G 磁珠】
* 准备工作:
* 配置Wash Buffer(含0.05% Tween20 的TBST):2.5 mL TBS(20×)用47.5 mL ddH2O稀释,再加入25μL Tween20,混匀,置于冰上 【注:TBS一般含有0.15 mol/L NaCl, 为减少非特异性结合,可以将NaCl增至 0.5mol/L】;
* 配置含PPI的ddH2O:ddH2O中加入十分之一体积的10× PPI(2mL:200μL),配置完后置于冰上;
* 配置1× SDS PAGE:5×SDS+10×PPI用RIPA(中)稀释;
* 平衡洗涤磁珠:
* 准备若干无酶离心管,标记;
* 轻轻混匀磁珠悬液后,为每个样品吸取25μL至1.5mL无酶EP管中;
* 每管加入175μL Wash Buffer,轻柔摇晃60s混匀;
* 放入磁力架静置60s,吸去上清,加入1mL Wash Buffer,轻柔摇晃60s混匀;
* 磁力架静置混匀,吸去上清;
* 将抗体-蛋白混合物加入清洗完的磁珠中,**4℃翻转孵育2-4h**,或摩天轮摇床室温孵育1h;
* 用磁力架分离结合有蛋白的磁珠,吸弃上清,加入500μL 配置好的Wash Buffer,混匀,磁力架分离。重复洗2遍;
* 加500μL 配置好的ddH2O,混匀洗涤1遍;
* 用磁力架分离磁珠,吸弃上清,加入50μL(说明书推荐100μL)**1× SDS** PAGE sample buffer,混匀;
* 95℃加热10min;
* 孵育期间标记新的EP管(包含日期,细胞名,样品名等);
* 用磁力架分离磁珠,吸取上清至新的EP管,-80℃保存。
### 【IP后的WB抗体选择】
* 如果IP和WB一抗来源(Host/Source)相同,IP后沉淀含有目的蛋白和IP抗体,在高温金属浴及还原剂的作用下,IP抗体发生发生变性,断裂为游离的重链(\~50kDa)和轻链(\~25kDa) 。后续做WB若使用前述的常规Anti-IgG(H+L)的酶标二抗,会同时识别原先的IP抗体和WB抗体,导致膜上至少出现三条不同的带。
* 解决方案一:WB一抗与IP一抗选择不同种属来源,此时选择普通二抗即可。
* 解决方案二:WB选用HRP偶联一抗,无需二抗。
* 解决方案三: 选用构象特异性或链型特异二抗,以避免重链和/或轻链的干扰。
* 构象特异性二抗,仅识别空间构象表位,高温变性的抗体失去了原有的空间构象,不会被识别,因此不会产生重链(\~50kDa)和轻链(\~25kDa)的条带。
* 当目标蛋白位于50kDa分子量附近时,可选择抗轻链的二抗,不识别重链,不会产生重链(\~50kDa)的条带。
* 当目标蛋白位于25kDa分子量附近时,可选择抗重链端的二抗,不识别轻链,不会产生轻链(\~25kDa)的条带。
### Day4:银染
* 将一半的IP样本用于银染,另一半可用于IP-MS/Co-IP。
* 用10孔梳以及1.0mm的玻璃板,配10或12.5%的胶,每孔加适量的蛋白样本,两边加适量marker(marker用1×SDS,1:3稀释),电泳至条带彻底分开。
* 配置固定液(依次加入50ml乙醇、10ml乙酸和40ml Milli-Q级纯水,混匀后即成100ml固定液),将膜置于固定液中(干净的玻璃器皿/15cm皿)固定过夜。
【注:用15cm皿则上述固定液体积及以下所有溶液体积减半】
【注:银染时戴口罩手套,不能用手或金属接触胶,用干净的塑料镊子在边缘小心地挪动凝胶】
* **30%乙醇洗涤:**\
弃固定液,加入100ml 30%乙醇,在摇床上室温摇动10分钟,摇动速度为60-70rpm。\
30%乙醇的配制:70ml Milli-Q级纯水或双蒸水中加入30ml乙醇,混匀后即成100ml 30%乙醇。
* **水洗涤:**\
弃30%乙醇,加入200ml Milli-Q级纯水或双蒸水,在摇床上室温摇动10分钟,摇动速度为60-70rpm。如果本步骤用水洗涤更长时间,对降低染色的背景略有帮助。
* **增敏:**\
弃水,加入100ml银染增敏液(1X),在摇床上室温摇动2分钟,摇动速度为60-70rpm。\
银染增敏液(1X)的配制:99ml Milli-Q级纯水或双蒸水中加入1ml银染增敏液(100X),混匀后即为银染增敏液(1X)。银染增敏液(1X)配制后需在2小时内使用。
* **水洗涤(共2次):**\
弃原有溶液,加入200ml Milli-Q级纯水或双蒸水,在摇床上室温摇动1分钟,摇动速度为60-70rpm。\
弃水,再加入200ml Milli-Q级纯水或双蒸水,在摇床上室温摇动1分钟,摇动速度为60-70rpm。
* **银染:**\
弃水,加入100ml银溶液(1X),在摇床上室温摇动10分钟,摇动速度为60-70rpm。\
银溶液(1X)的配制:99ml Milli-Q级纯水或双蒸水中加入1ml银溶液(100X),混匀后即为银溶液(1X)。银溶液(1X)配制后需在2小时内使用。
* **水洗涤:**\
弃原有溶液,加入100ml Milli-Q级纯水或双蒸水,在摇床上室温摇动1-1.5分钟,摇动速度为60-70rpm。\
注意:水洗涤的时间不能超过1-1.5分钟。
* **显色:**\
弃水,加入100ml银染显色液,在摇床上室温摇动3-10分钟,直至出现比较理想的预期蛋白条带,摇动速度为60-70rpm。\
银染显色液的配制:80ml Milli-Q级纯水或双蒸水中加入20ml银染基本显色液(5X),再加入0.05ml银染显色加速液(2000X),混匀后即为银染显色液。银染显色液配制后需在20分钟内使用。
* **终止:**\
弃银染显色液,加入100ml银染终止液(1X),在摇床上室温摇动10分钟,摇动速度为60-70rpm。终止时有气体产生属正常现象,产生的气体为二氧化碳。\
银染终止液(1X)的配制:95ml Milli-Q级纯水或双蒸水中加入5ml银染终止液(20X),混匀后即为银染终止液(1X)。银染终止液(1X)配制后宜当天使用。
* **水洗涤:**\
弃银染终止液,加入100ml Milli-Q级纯水或双蒸水,在摇床上室温摇动2-5分钟,摇动速度为60-70rpm。
* **保存:**\
可在Milli-Q级纯水或双蒸水中保存。或采用适当的方式制备成干胶。
* 拍摄:
用手机或者Biorad显像系统(白色底板)拍摄银染图片。
### IP-MS送样
* 配置1.0mm的10孔胶,每孔加25μL左右蛋白,左右加marker,每条泳道之间也用marker隔开;
* 电泳至下层胶1-2cm之内,切割胶条,置于EP管保存。
如捕获标签-融合蛋白,可以选用纳米抗体,即羊驼重链抗体可变区(VHH),使用普通二抗也可以避免轻重链干扰。
## 实验步骤
### 前期准备
* 在2个T75或者2个10cm皿内铺板293T,使用PEI转染质粒,等待2天;
### Day1:收集、裂解细胞
* 预冷PBS、EP管,取冰若干;
* 提前配置蛋白裂解液:每组样本准备1000μL的RIPA(中)+10μL的PMSF(100×)/100μL的PPI(10×),置于冰上。
* 吸出两个瓶/皿中的培养液,加1mL预冷的1×PBS,再用细胞刮刀将细胞刮下,将细胞转入预冷的EP管中,4℃,500G离心5min,弃去上清,用预冷的1X PBS重悬细胞,离心,弃上清,重复两次。
* 使用1000μL裂解液重悬细胞。
* 将EP管放于冰上30min,每隔10min重悬细胞一次。
* 摩天轮 4°C 转动 40-60min 以彻底裂解细胞(摩天轮上ep管可以用封口膜封口);
* 将蛋白冻存于-80℃,或直接进行下一步;
### Day2:平衡磁珠、结合蛋白
### 【康体FLAG磁珠】
* 取出蛋白,在冰上融化,12000G离心15min;
* 吸出50μL作为input,向其中加入12μL 5×SDS,混匀,置于-80℃;
* 配置1×PBST:1×PBS中加入0.05%的Tween20(50mL:25μL),配置完后置于冰上;
* 涡旋Anti-Flag Magarose Beads磁珠,吸取25μL置于1.5mL 无酶EP管中;
* 加入500μL 1XPBST(1X PBS+0.05%Tween-20),手动摇晃60s;
* 放入磁力架静置60s,吸去上清,再加入500μL 1XPBST,随后在4℃摩天轮转动5min;再次放入磁力架吸附,重复3次;
* 在平衡好的磁珠中加入细胞蛋白提取液,用封口膜封口后4℃摇床翻转过夜;
{% file src="" %}
### 【Thermo FLAG磁珠】
* 准备工作:
* 打开95℃金属浴;
* 准备RIPA(中)、 5×SDS、1×PBS以及10×PPI,以及若干EP管,置于冰上;
* 取出蛋白样品,在冰上融化,12000G离心15min;
* 取出磁珠,使用前将磁珠平衡至室温;
* 离心期间平衡洗涤磁珠、标记无酶离心管:
* 配置含PPI的裂解液(Binding Buffer):RIPA(中)加入十分之一体积的10× PPI(10mL:1mL),配置完后置于冰上;
* 涡旋或吹匀Anti-Flag Magnetic Agarose磁性琼脂糖珠,为每个样品吸取25μL(说明书推荐50μL)置于1.5mL 无酶EP管中;
* 每管加入500μL 上述配置的裂解液,手动摇晃60s混匀;
* 放入磁力架静置60s,吸去上清,加入500μL上述裂解液重悬,随后在4℃摩天轮转动5min;
* 再次放入磁力架吸附,弃上清,加入500μL裂解液,重悬洗涤5min,再磁力架吸附,弃去上清;
* 从离心完的蛋白样品上清中吸出50μL作为input,向其中加入12μL 5×SDS,混匀,95℃加热10min,置于-80℃;
* 其余样品加入到平衡好的磁珠管中,再加入10× PPI,用封口膜封口后4℃摇床翻转过夜;
{% file src="" %}
### 【Thermo Protein A/G 磁珠】
* 准备工作:
* 提前打开95℃金属浴;
* 准备5× SDS以及若干EP管(蛋白上清若干管,input若干管,磁珠若干管);
* 取出蛋白样品,在冰上融化,12000G离心15min;
* 从离心完的蛋白样品上清中吸出50μL作为input,向其中加入12μL **5×SDS**,混匀,95℃加热10min,置于-80℃;
* 将其余**蛋白上清**分为2份,分别与约10μL的IgG同型对照抗体/目的蛋白**抗体**进行混合(每个T75的293T约1.2mg蛋白,抗体浓度一般1-5 µL/mg;2个T75的蛋白量即对应2-10μL抗体),**4℃翻转过夜**(若阴性对照有阳性结果,可以减少孵育时间至室温孵育1-2h);
{% file src="" %}
### Day3:洗涤、洗脱
### 【康体FLAG磁珠】
* 用磁力架静置分离磁珠1min,吸去上清液;
* 加入500μL PBST悬浮磁珠,4℃冰箱旋转摇晃5min;再静置,如此重复洗涤4次;
* 配置30μL 1× SDS loading buffer,加入PMSF/PPI;
* 吸去上清液,用30μL **1×SDS** loading重悬磁珠;
* 95℃加热磁珠10min,使免疫沉淀复合物与磁珠分离;
* 用磁力架静置分离磁珠,取上清,冻于-80℃,跑SDS-PAGE。
### 【Thermo FLAG磁珠】
* 准备工作:
* 提前打开95℃金属浴;
* 配置含PPI的1×PBS(Wash Buffer):1×PBS中加入十分之一体积的10× PPI(10mL:1mL),配置完后置于冰上;
* 配置含PPI的ddH2O:ddH2O中加入十分之一体积的10× PPI(2mL:200μL),配置完后置于冰上;
* 配置1× SDS PAGE sample buffer:5×SDS+10×PPI用RIPA(中)稀释;
* 用磁力架分离磁珠,吸弃上清,加入500μL 配置好的Wash Buffer,混匀,磁力架分离。重复洗一遍;
* 加500μL 配置好的ddH2O,混匀洗涤1遍;
* 用磁力架分离磁珠,吸弃上清,加入50μL(说明书推荐100μL)**1× SDS PAGE** sample buffer,混匀;
* 95-100℃加热10分钟(期间标记EP管);
* 用磁力架分离磁珠,吸取上清至新的EP管,-80℃保存。
### 【Thermo Protein A/G 磁珠】
* 准备工作:
* 配置Wash Buffer(含0.05% Tween20 的TBST):2.5 mL TBS(20×)用47.5 mL ddH2O稀释,再加入25μL Tween20,混匀,置于冰上 【注:TBS一般含有0.15 mol/L NaCl, 为减少非特异性结合,可以将NaCl增至 0.5mol/L】;
* 配置含PPI的ddH2O:ddH2O中加入十分之一体积的10× PPI(2mL:200μL),配置完后置于冰上;
* 配置1× SDS PAGE:5×SDS+10×PPI用RIPA(中)稀释;
* 平衡洗涤磁珠:
* 准备若干无酶离心管,标记;
* 轻轻混匀磁珠悬液后,为每个样品吸取25μL至1.5mL无酶EP管中;
* 每管加入175μL Wash Buffer,轻柔摇晃60s混匀;
* 放入磁力架静置60s,吸去上清,加入1mL Wash Buffer,轻柔摇晃60s混匀;
* 磁力架静置混匀,吸去上清;
* 将抗体-蛋白混合物加入清洗完的磁珠中,**4℃翻转孵育2-4h**,或摩天轮摇床室温孵育1h;
* 用磁力架分离结合有蛋白的磁珠,吸弃上清,加入500μL 配置好的Wash Buffer,混匀,磁力架分离。重复洗2遍;
* 加500μL 配置好的ddH2O,混匀洗涤1遍;
* 用磁力架分离磁珠,吸弃上清,加入50μL(说明书推荐100μL)**1× SDS** PAGE sample buffer,混匀;
* 95℃加热10min;
* 孵育期间标记新的EP管(包含日期,细胞名,样品名等);
* 用磁力架分离磁珠,吸取上清至新的EP管,-80℃保存。
### 【IP后的WB抗体选择】
* 如果IP和WB一抗来源(Host/Source)相同,IP后沉淀含有目的蛋白和IP抗体,在高温金属浴及还原剂的作用下,IP抗体发生发生变性,断裂为游离的重链(\~50kDa)和轻链(\~25kDa) 。后续做WB若使用前述的常规Anti-IgG(H+L)的酶标二抗,会同时识别原先的IP抗体和WB抗体,导致膜上至少出现三条不同的带。
* 解决方案一:WB一抗与IP一抗选择不同种属来源,此时选择普通二抗即可。
* 解决方案二:WB选用HRP偶联一抗,无需二抗。
* 解决方案三: 选用构象特异性或链型特异二抗,以避免重链和/或轻链的干扰。
* 构象特异性二抗,仅识别空间构象表位,高温变性的抗体失去了原有的空间构象,不会被识别,因此不会产生重链(\~50kDa)和轻链(\~25kDa)的条带。
* 当目标蛋白位于50kDa分子量附近时,可选择抗轻链的二抗,不识别重链,不会产生重链(\~50kDa)的条带。
* 当目标蛋白位于25kDa分子量附近时,可选择抗重链端的二抗,不识别轻链,不会产生轻链(\~25kDa)的条带。
### Day4:银染
* 将一半的IP样本用于银染,另一半可用于IP-MS/Co-IP。
* 用10孔梳以及1.0mm的玻璃板,配10或12.5%的胶,每孔加适量的蛋白样本,两边加适量marker(marker用1×SDS,1:3稀释),电泳至条带彻底分开。
* 配置固定液(依次加入50ml乙醇、10ml乙酸和40ml Milli-Q级纯水,混匀后即成100ml固定液),将膜置于固定液中(干净的玻璃器皿/15cm皿)固定过夜。
【注:用15cm皿则上述固定液体积及以下所有溶液体积减半】
【注:银染时戴口罩手套,不能用手或金属接触胶,用干净的塑料镊子在边缘小心地挪动凝胶】
* **30%乙醇洗涤:**\
弃固定液,加入100ml 30%乙醇,在摇床上室温摇动10分钟,摇动速度为60-70rpm。\
30%乙醇的配制:70ml Milli-Q级纯水或双蒸水中加入30ml乙醇,混匀后即成100ml 30%乙醇。
* **水洗涤:**\
弃30%乙醇,加入200ml Milli-Q级纯水或双蒸水,在摇床上室温摇动10分钟,摇动速度为60-70rpm。如果本步骤用水洗涤更长时间,对降低染色的背景略有帮助。
* **增敏:**\
弃水,加入100ml银染增敏液(1X),在摇床上室温摇动2分钟,摇动速度为60-70rpm。\
银染增敏液(1X)的配制:99ml Milli-Q级纯水或双蒸水中加入1ml银染增敏液(100X),混匀后即为银染增敏液(1X)。银染增敏液(1X)配制后需在2小时内使用。
* **水洗涤(共2次):**\
弃原有溶液,加入200ml Milli-Q级纯水或双蒸水,在摇床上室温摇动1分钟,摇动速度为60-70rpm。\
弃水,再加入200ml Milli-Q级纯水或双蒸水,在摇床上室温摇动1分钟,摇动速度为60-70rpm。
* **银染:**\
弃水,加入100ml银溶液(1X),在摇床上室温摇动10分钟,摇动速度为60-70rpm。\
银溶液(1X)的配制:99ml Milli-Q级纯水或双蒸水中加入1ml银溶液(100X),混匀后即为银溶液(1X)。银溶液(1X)配制后需在2小时内使用。
* **水洗涤:**\
弃原有溶液,加入100ml Milli-Q级纯水或双蒸水,在摇床上室温摇动1-1.5分钟,摇动速度为60-70rpm。\
注意:水洗涤的时间不能超过1-1.5分钟。
* **显色:**\
弃水,加入100ml银染显色液,在摇床上室温摇动3-10分钟,直至出现比较理想的预期蛋白条带,摇动速度为60-70rpm。\
银染显色液的配制:80ml Milli-Q级纯水或双蒸水中加入20ml银染基本显色液(5X),再加入0.05ml银染显色加速液(2000X),混匀后即为银染显色液。银染显色液配制后需在20分钟内使用。
* **终止:**\
弃银染显色液,加入100ml银染终止液(1X),在摇床上室温摇动10分钟,摇动速度为60-70rpm。终止时有气体产生属正常现象,产生的气体为二氧化碳。\
银染终止液(1X)的配制:95ml Milli-Q级纯水或双蒸水中加入5ml银染终止液(20X),混匀后即为银染终止液(1X)。银染终止液(1X)配制后宜当天使用。
* **水洗涤:**\
弃银染终止液,加入100ml Milli-Q级纯水或双蒸水,在摇床上室温摇动2-5分钟,摇动速度为60-70rpm。
* **保存:**\
可在Milli-Q级纯水或双蒸水中保存。或采用适当的方式制备成干胶。
* 拍摄:
用手机或者Biorad显像系统(白色底板)拍摄银染图片。
### IP-MS送样
* 配置1.0mm的10孔胶,每孔加25μL左右蛋白,左右加marker,每条泳道之间也用marker隔开;
* 电泳至下层胶1-2cm之内,切割胶条,置于EP管保存。